GATK4:germline SNP calling
1. 简介
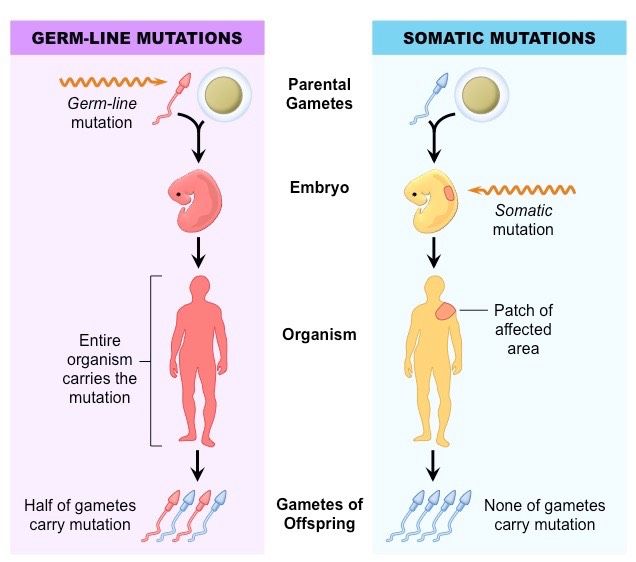
单核苷酸多态性(single nucleotide polymorphism,SNP),主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。从来源上看,SNP可以分为 “germline SNP” 和 “somatic SNP”。
如下图所示,Germline的mutation其实来自于上一代,这种mutation会随着个体整个胚胎发育过程存在。在研究方法上,Germline的mutation在家系分析中占有重要角色,对很多遗传病(其中包括遗传型肿瘤)的研究中占有重要地位。在测序过程中,除了个体测序之外,还会加上个体的家属一同测序。病理分析时也会考虑家族背景的影响。
somatic突变是不遗传的,在研究方法上主要偏重采集癌症组织和正常组织进行比较得到结果。因此,在call somatic mutation的时候最好有同一个体的正常组织进行参照。从研究意义来讲,somatic更侧重于单个患者的癌症分型和发病机理研究。
2. GATK4下载安装
在GATK的官方网站下载目前最新版本4.1.2.0。并将软件压缩文件上传至Linux服务器系统上。解压缩即可使用大部分功能,但涉及到germline CNV calling的数据分析则需要安装相应的python环境。
3. 准备相关数据:所有准备的数据都保存在一个index的文件目录下
3.1 下载相关数据
这里以分析人类的数据作为案例。选择的参考基因组为hg38,相应的数据由broadinstitute提供:
1 | lftp ftp.broadinstitute.org/bundle -u gsapubftp-anonymous #没有密码 |
进入该ftp后至少需要下载如下的数据:
- Homo_sapiens_assembly38.fasta.gz
- 1000G_phase1.snps.high_confidence.hg38.vcf.gz
- 1000G_phase1.snps.high_confidence.hg38.vcf.gz.tbi
- dbsnp_146.hg38.vcf.gz
- dbsnp_146.hg38.vcf.gz.tbi
- Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
- Mills_and_1000G_gold_standard.indels.hg38.vcf.gz.tbi
3.2 建立索引文件
- 通过
BWA软件,为参考基因组hg38建立索引文件:1
bwa index hg38.fa
- 通过samtools软件,为参考基因组 hg38 建立fai索引文件:
1
samtools faidx hg38.fa
4. 数据分析流程
4.1 质控
通过
FastQC软件检测二代测序原始数据,如果数据已经是被清理过的干净数据,则可以直接进行比对。如果数据不是干净的数据,则需要进行数据清理步骤,即使用
Trimmomatic等软件进行数据清理。
通过质控获得的是一个比较干净的测序原始数据(cleandata.fq)。
4.2 比对
- 通过软件
BWA将干净的测序数据比对到参考基因组上:
1 | bwa mem -t 15 -M -R "@RG\tID:G118bloodEX\tSM:G118bloodEX\tLB:WES\tPL:Illumina" /GPFS/liangbilin/WES/index/Homo_sapiens.GRCh38.dna.chromosome.fa G118bloodEX_1.fastq G118bloodEX_2.fastq 1> ../aln/G118bloodEX.sam 2> ../aln/G118bloodEX.log |
-t 设置软件线程数
-M 用来处理同一个reads比对到参考基因组上不同位置的情况。官方解释为:mark shorter split hits as secondary. 设置该参数的目的是为了兼容GATK的markDuplicates模块。
-R 接的是 Read Group的字符串信息,它是用来将比对的read进行分组的,这个信息对于我们后续分析非常重要。具体分析时候需要修改该参数的信息。
- 把sam格式转为bam格式
1 | samtools view -bS G118bloodEX.sam > G118bloodEX.bam |
- 对bam文件进行排序
1 | samtools sort -@ 20 G118bloodEX.bam -o G118bloodEX.sorted.bam |
4.3 标记重复
在NGS测序之前都需要先构建测序文库:通过物理(超声)打断或者化学试剂(酶切)切断原始的DNA序列,然后选择特定长度范围的序列去进行PCR扩增并上机测序。这个过程中产生的重复reads,增大了变异检测结果的假阴率和假阳率:
- PCR反应过程中也会带来新的碱基错误。发生在前几轮的PCR扩增发生的错误会在后续的PCR过程中扩大,同样带来假的变异;
- PCR反应可能会对包含某一个碱基的DNA模版扩增更加剧烈(这个现象称为PCR Bias);
- 如果某个变异位点的变异碱基都是来自于PCR重复,而我们却认为它深度足够判断是真的变异位点,这个结论其实有很大可能是假阳性。
通过GATK4的MarkDuplicates标记重复:
1 | gatk MarkDuplicates -I G118bloodEX.sorted.bam -O G118bloodEX.sorted.marked.bam -M G118bloodEX.metrics |
4.4 矫正bam文件
检测并矫正双端测序的mate信息。如果上一步骤只是标记重复而不是删除,理论上不需要进行该步骤。
1 | gatk FixMateInformation -I G118bloodEX.sorted.marked.bam -O G118bloodEX.sorted.marked.fixed.bam -SO coordinate |
4.5 为bam文件生成索引
1 | samtools index G118bloodEX.sorted.marked.fixed.bam |
4.6 调整碱基测序质量值
变异检测是一个极度依赖测序碱基质量值,因为这个质量值是衡量我们测序出来的这个碱基到底有多正确的重要指标。对base的quality score进行校正)碱基质量分数重校准(Base quality score recalibration,BQSR),就是利用机器学习的方式调整原始碱基的质量分数。它分为两个步骤:
- 利用已有的snp数据库,建立相关性模型,产生重校准表
1 | gatk BaseRecalibrator -R /GPFS/liangbilin/biomarker/index/Homo_sapiens_assembly38.fa -I G118bloodEX.sorted.marked.fixed.bam --known-sites /GPFS/liangbilin/biomarker/index/dbsnp_146.hg38.vcf.gz --known-sites /GPFS/liangbilin/biomarker/index/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz -O G118bloodEX.recal.table |
-R 参考基因组
-I 输入的bam文件
–known-sites 已知的变异位点文件,同时需要已经创建索引文件
-O 输出重校准表
- 根据这个模型对原始碱基进行调整,只会调整非已知SNP区域。
1 | gatk ApplyBQSR -R /GPFS/liangbilin/biomarker/index/Homo_sapiens_assembly38.fa -I G118bloodEX.sorted.marked.fixed.bam -bqsr G118bloodEX.recal.table -O G118bloodEX.sorted.marked.fixed.bqsr.bam |
4.7 变异检测前确定bam文件是否符合GATK要求 (选做)
运行GATK4的 ValidateSamFile 模块,如果显示 No Error,则可以用HaplotypeCaller call SNP/Indel。
1 | ~/program/gatk-4.1.2.0/gatk ValidateSamFile -I G118bloodEX.sorted.marked.fixed.bqsr.bam >>G118bloodEX.ValidateSamFile.log 2>&1 |
4.8 Call candidate variants
对于做 germline 情况,利用 HaplotypeCaller 检测突变。HaplotypeCaller的应用有两种做法,区别在于是否生成中间文件gVCF:
- 直接进行HaplotypeCaller,这适合于单样本,只执行一次HaplotypeCaller。
1 | gatk HaplotypeCaller -R /GPFS/liangbilin/biomarker/index/Homo_sapiens_assembly38.fa -I G118bloodEX.sorted.marked.fixed.bqsr.bam -D ../index/dbsnp_146.hg38.vcf.gz -A QualByDepth -A RMSMappingQuality -A MappingQualityRankSumTest -A ReadPosRankSumTest -A FisherStrand -A StrandOddsRatio -A Coverage -O G118bloodEX.vcf |
-I 准备好的BAM文件,可以提交多个( -I 1.bam -I 2.bam -I 3.bam … )
- 如果多样本,每增加一个样本数据都需要重新运行这个HaplotypeCaller,而这个时候算法需要重新去读取所有人的BAM文件,浪费大量时间。每个样本先各自生成gVCF,然后再进行群体joint-genotype。gVCF全称是genome VCF,是每个样本用于变异检测的中间文件,格式类似于VCF,它把joint-genotype过程中所需的所有信息都记录在这里面,文件无论是大小还是数据量都远远小于原来的BAM文件。这样一旦新增加样本也不需要再重新去读取所有人的BAM文件了,只需为新样本生成一份gVCF,然后重新执行这个joint-genotype就行了。
1 |
原文作者: Billy & Barney
原文链接: https://liangbilin.github.io/2019/12/01/Billy--GATK4—germline SNP calling/
版权声明: 转载请注明出处(必须保留作者署名及链接)