Nanopore Sequencing 概述
为什么要选择 Nanopore 测序?
(一) 超长读长
在纳米孔测序中,读长长度可以等于输入片段长度。读长长度不受限于测序设备,用户可以通过所使用的文库制备实验方案来控制片段长度。目前报到处DNA片段长度最高记录为>2 Mb,直接RNA测序读长最长为26kb。长读长提供了一个更明确的方法来比对和匹配DNA或RNA序列,提供高质量、更完整、更连续的基因组组装。在类如植物基因组和具有大型结构变异和高水平重复区域的基因组中优势显著。
(二) 直接测序
纳米孔技术基于电子学原理,允许直接测序原始DNA和RNA。不需要通过DNA拷贝、进行链合成或使用亚硝酸盐进行转化,这不仅节省了时间和成本,还意味着碱基修饰的信息(例如5mC,pseudouridine,m6A)会被完整的保留,并且包含在测序运行产生的原始信号信息中,随时进行分析。由于纳米孔技术支持无需PCR的直接测序,也就没有了扩增偏好性,并且文库制备工作流程也更简单。
(三) 实时测序
与在运行结束时批量交付数据的传统测序技术不同,纳米孔技术提供的是动态、实时的测序,支持在几分钟内就获得病原体鉴定等时间关键型应用的检测结果。测序时,DNA快速通过纳米孔。DNA片段通过纳米孔的速率已从推出时的每秒35个碱基提升到了现在每秒450个碱基。每一个在阵列上完成的读长,在数秒后就可以用来开始数据分析,而不是几小时甚至几天。用户可以在测序早期了解样本的质量和状态,也可以在获得足够的数据后停止测序。快速的采样到结果周转时间在未来感染性疾病、实地动态监测疫情爆发以及其它诊断方面具有巨大潜力。
(四) 按需要测序
与传统测序技术不同的是,在纳米孔测序中,用户可以自行掌握测序时间、地点、以及需要使用的芯片数量。例如,可以单次使用的最小型的测序芯片Flongle适合快速质量检测、小型基因组实验和靶向测序。便携式测序仪MinION,重量不足100g,可以被带到任何地方测序任何样本。台式测序仪GridION和PromethION分别具有5个和24或者48个测序芯片,每个芯片可以独立运行,也可以同时使用,并结合实时的数据传送。这意味着用户可以根据样本的数量选择芯片数量,随数据量要求随时启动和停止实验。
(五) 启动费用低
传统的高通量测序价格昂贵,启动需要数十万至百万人民币。而纳米孔测序目前启动套装只需要17271元,包括MinION测序仪,两张测序芯片,一盒建库试剂盒,一盒清洗试剂盒。两张芯片最多可以测序60G数据。
(六) 灵活、可扩展
所有Oxford Nanopore测序设备使用同样的核心技术,用户可以轻松根据应用测试实验以及扩大或缩小规模。从最小型的Flongle和掌上MinION,到桌面型的GridION和PromethION。所有设备都可用于进行按需测序实验。包括从单次试验到超高通量项目,全部都可提供快速、长读长、实时的DNA或RNA直接测序。用于纳米孔测序的DNA或RNA建库过程简单直接,最快的建库方法只需5到10分钟就可在样本分子末端添加测序接头及马达蛋白。
Nanopore 测序的典型应用
(一) 大基因组拼接
nanopore最显著特点就是读长长。长读长对于大基因组的拼接将会产生立竿见影的效果。在以往基于短片段的基因组拼接中,由于一些动植物基因组本身具有多倍体,高度重复,高度杂合的特性,导致基因组拼接是一项异常艰难的工作,有些植物甚至复杂到利用短片段根本无法完成拼接工作,例如基因组大小高达152Gb的重楼百合 (Paris japonica),还有10倍体的择捉草莓 (Fragaria turupensis)等。虽然后来有了大片段文库,BAC文库,optical mapping光学图谱,Hi-C,bionano等辅助技术,但依然无法从根本上解决大型基因组拼接的问题。而nanopore测序技术将从根本上改变大型基因组拼接技术难题,根据百迈克公司公开发布的数据来看。利用三代测序可以极大的提高基因组的完整性contig数目减少,N50长度可以达到M级别,同时配合多轮纠错机制,拼接的准确性也非常高。
(二) 细菌完成图
由于细菌基因组比较小,通常小于10M,大部分是一条染色体,并且由于细菌基因组通常重复序列不多,利用nanopore的长读长的特性,甚至可以一次性拼接出完整的基因组。笔者以前做过上千微生物基因组拼接,当时需要利用短片段多文库的策略拼接细菌完整图,折腾一个多月也无法彻底解决大的重序列的问题,而现在利用nannopor测序,只需几分钟(根据具体样品和硬件配置不同)就可以拼出一条完整的基因组(我们后面会有具体的推文)。
(三) 全长无偏倚转录组
以往的转录组分析,由于无法直接对RNA进行测序,往往需要先对mRNA进行打断,在反转录为cDNA。反转录过程中PCR可能会引入扩增偏差,导致筛选到假阳性的表达差异基因,尽管目前采用单细胞加UMI特异标签的方法减少PCR偏差。但依然无法获取和分析全长转录本,由于真核生物普遍存在的剪切差异,短读长测序依然无法准确识别,而利用nanopore的长读长测序,可以准确识别各基因的多个同源异构体,简单准确。并且nanopore可以对RNA直接测序,这样就能够直接识别RNA的碱基修饰。
(四) 大片段结构变异
目前的基因组突变分析主要在单碱基突变SNP,以及少量的插入缺失(InDel)上,而忽略掉基因组上含量巨大的大的结构变异。这主要是因为仅使用短测序读长时,无法准确检测这些变异,例如缺失、插入、重复、倒位和易位。但已有研究表明,这些重复区域和结构变异与人类的健康和疾病有关(例如,老龄化、三联体扩张疾病、自闭症、癫痫和癌症),使得对它们进行的常规表征鉴定非常有利。利用nanopore长读长的特点,非常适合进行大片段结构变异的检测。
(五) 突变定相分析
所谓定相分析,被称为Phasing,简单来说就是将一个二倍体(甚至是多倍体)基因组上的等位基因信息,判断出其遗传自父本还是母本,最终使得所有来自同一个亲本的信息都能够排列在同一条染色体里面。很显然,测序的长度越长对定相分析越有利(如果是完整一条就直接比对就行了)。根据文献报道,利用超长读长不仅使组装连续性提高了一倍(NG50~6.4MB),而且显著改善了定相(phasing)等位基因的功能。例如,有可能对包含在单个16 MB contig中的整个4 MB主要组织相容性复合体(MHC)进行定相(phasing)。
(六) 微生物快速鉴定
利用nanopore测序,实时,快速的特点,非常适合为微生物的快速鉴定。可以在采集点分析直接进行测序,而且实时的basecalling,上机几分钟之后就可以有序列产出,可以直接使用序列进行物种分类鉴定。从样本到检测结果用时大大减少。据报道,使用MinION在北极环境中从样本采集到测序数据生成仅需不到40小时。
Nanopore 测序原理
Nanopore 测序是将人工合成的一种多聚合物的膜浸在离子溶液中,多聚合物膜上布满了经改造的穿膜孔的跨膜通道蛋白(纳米孔),也就是Reader蛋白。在膜两侧施加不同的电压产生电压差,DNA链在马达蛋白的牵引下,解螺旋通过纳米孔蛋白,不同的碱基会形成特征性离子电流变化信号。该膜具有非常高的电阻。通过对浸在电化学溶液中的膜上施加电势,可以通过纳米孔产生离子电流。进入纳米孔的单分子引起特征性的电流干扰,这被称为Nanopore信号。
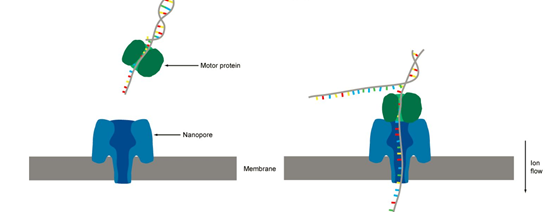
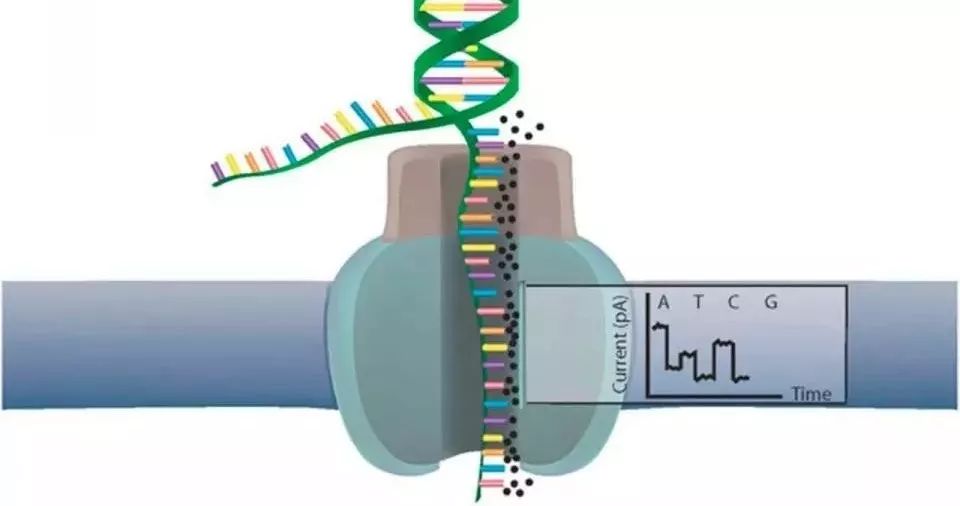
Nanopore 测序数据格式
Nanopore 测序时需要使用一款名为 MinKNOW 的软件。MinKNOW 是 Nanopore 测序所有平台的驱动操作软件,主要功能包括控制仪器,测序与实时碱基识别,检测运行状态,数据采集。在运行之前,可以选择测序输出文件是 FAST5格式 还是 FASTQ格式 。
FASTQ 格式是一种带有碱基和质量值的序列文件,目前是测序行业非常标准的文件格式。目前作为illumina,Ion Torrent,BGIseq,Pacbio,nanopore等平台通用的测序文件表示形式,这样后续很多软件可以通用。
FAST5 格式存储了Nanopore测序过程中全部的输出信息。里面记录着设备运行时全部的信息,包括捕获的电信号值,设备运行时间,电压,温度等等信息。
FAST5 格式
FAST5是HDF5文件格式的一个变种,而HDF(Hierarchical Data Format),是一种设计用于存储和组织大量数据的文件格式,一般扩展名为.hdf5或.h5,表示现在使用的版本是第五个版本。这是一种分级的数据文件,可以存储不同类型的图像和数码数据的文件格式,并且可以在不同类型的机器上传输,同时还有统一处理这种文件格式的函数库。fast5里面可以包含很多的内容,并且可以继续添加。简单理解,这种文件类似于一个经过压缩的文件夹,里面包含很多文件,如下图所示:
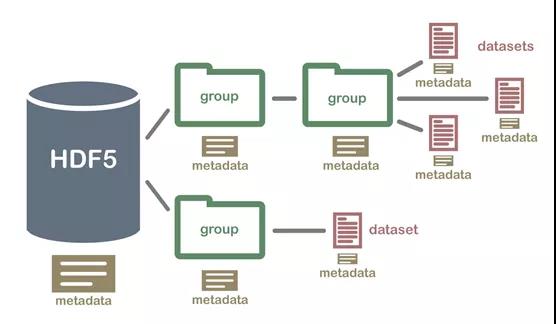
nanopore测序过程中,一个纳米孔测序完一条序列,则可以输出一个fast5文件,一个MinION的flowcell就生成10-20Gb的数据量,有几百万的条reads,这百万千万级别的文件处理就是一个大麻烦,需要反复打开关闭IO,完成一次文件拷贝和统计要花很长时间。19年以前的fast5文件是一条序列一个fast5文件,现在新的MinKNOW做了升级,可以设定生成multi_fast5文件,一个fast5包含的reads数目可以自己设定,一般4,000。
如何查看fast5格式文件
可以使用软件 HDFView 查看FAST5文件格式。 https://www.hdfgroup.org/downloads/hdfview/
FAST5格式文件的优缺点
FAST5格式的优点是内容非常全,可以存储所有的信息。缺点也非常明显,就是占用空间特别大。例如,23M左右的碱基序列,存储为fastq格式大概45M,压缩之后大约是23M;而原始的fast5文件则需要占用613M的存储,大约30倍。如果是测序一个MiniION 30G的碱基,则大概需要将近1T的存储。
拆分与合并fast5格式
有时候,一条测序读长被保存在一个FAST5文件,有时候一个FAST5文件可能保存了多条读长。
1 | #ont_fast5_api 网址 |
使用案例
1 | #多条读长合并为一个文件 |
原文作者: Billy & Barney
原文链接: https://liangbilin.github.io/2019/10/02/Billy--Nanopore Sequencing 概述/
版权声明: 转载请注明出处(必须保留作者署名及链接)