转录组差异表达分析--免比对工具kallisto
(一) RNA-seq概述
RNA-seq是研究转录组应用最广泛,也是最重要的技术之一。RNA-seq分析内容包括序列比对、转录本拼接、表达定量、差异分析、融合基因检测、可变剪接、RNA编辑和突变检测等,具体流程和常用工具如下图所示。通常的分析不一定需要走完全部流程,按需进行,某些步骤可以跳过、简化等。
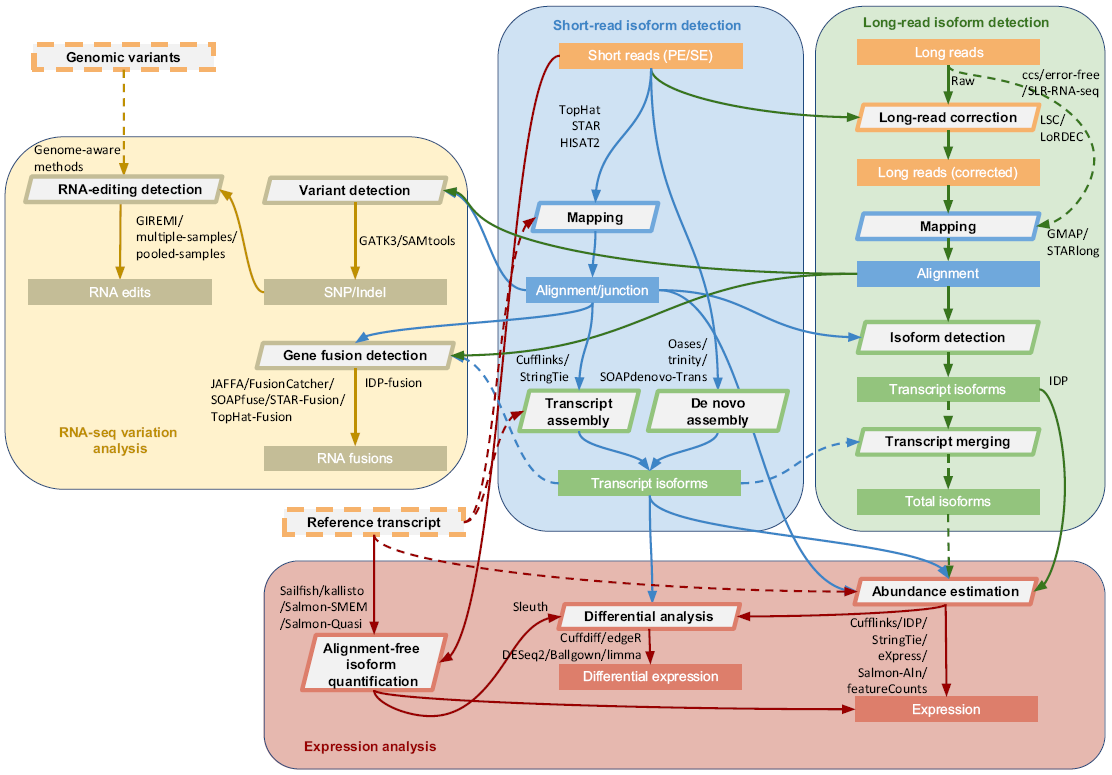
RNA-seq中最常用的分析方法就是找出差异表达基因(Differential gene expression, DEG)。在实验室中,标准流程就分为三步:
step1:构建测序文库。包括提取RNA,富集mRNA或清除核糖体RNA,合成cDNA,加上接头。
step2:在高通量测序平台(通常为Illumina)上对文库进行测序,每个样本的测序深度为10-30M读长。
step3:数据分析。具体而言:对测序得到的读长进行比对或组装到转录本上;对覆盖到每个基因区域的读长进行计数;根据统计模型鉴定不同样本间差异表达的基因。(这种分析过程是比较传统的方法)
(二) 差异表达分析——免比对工具kallisto
2016年4月4日,Nature Biotechnology 杂志上发表了一篇题为“Near-optimal probabilistic RNA-seq quantification”的论文,主要介绍一个RNA-seq数据量化软件kallisto。传统的RNA-seq数据分析思路需要将测序数据先比对到参考基因组上(tophat2、bowtie2、HISAT2等软件),然后从比对结果中对覆盖到每个基因区域的读长进行计数并计算表达量。
这篇论文介绍的kallisto最大的优势在于保证了量化分析准确性的前提下,去掉了比对步骤,极大降低了计算资源的需求。而它的原理是基于一个被证明的理论,即read具体比对到参考基因的什么位置并不影响最终表达量的计算,换句话说只需要确定这个read属于该基因即可,具体它在基因的哪个区域并不重要。
(三) 简明教程
安装kallisto
通过Bioconda安装:
1 | # 创建虚拟环境 |
构造索引文件
- 在ensembl、UCSC或NCBI网站上可下载对应物种的FASTA格式的
cDNA文件。 - 使用
kallisto软件对物种的cDNA文件构建索引文件:
1 | # 选择kallisto的index模块 |
基因表达定量
双端测序数据分析过程:
1 | # 选择kallisto的quant模块 |
单端测序数据分析过程:
1 | kallisto quant -i hg38.idx -o output --single -l 200 -s 20 test1.fq.gz |
(四) 参考资料
原文作者: Billy & Barney
原文链接: https://liangbilin.github.io/2019/09/07/Billy--转录组差异表达分析——kallisto/
版权声明: 转载请注明出处(必须保留作者署名及链接)