转录组差异表达分析概述
(一) RNA-seq概述
RNA-seq是研究转录组应用最广泛,也是最重要的技术之一。RNA-seq分析内容包括序列比对、转录本拼接、表达定量、差异分析、融合基因检测、可变剪接、RNA编辑和突变检测等,具体流程和常用工具如下图所示。通常的分析不一定需要走完全部流程,按需进行,某些步骤可以跳过、简化等。
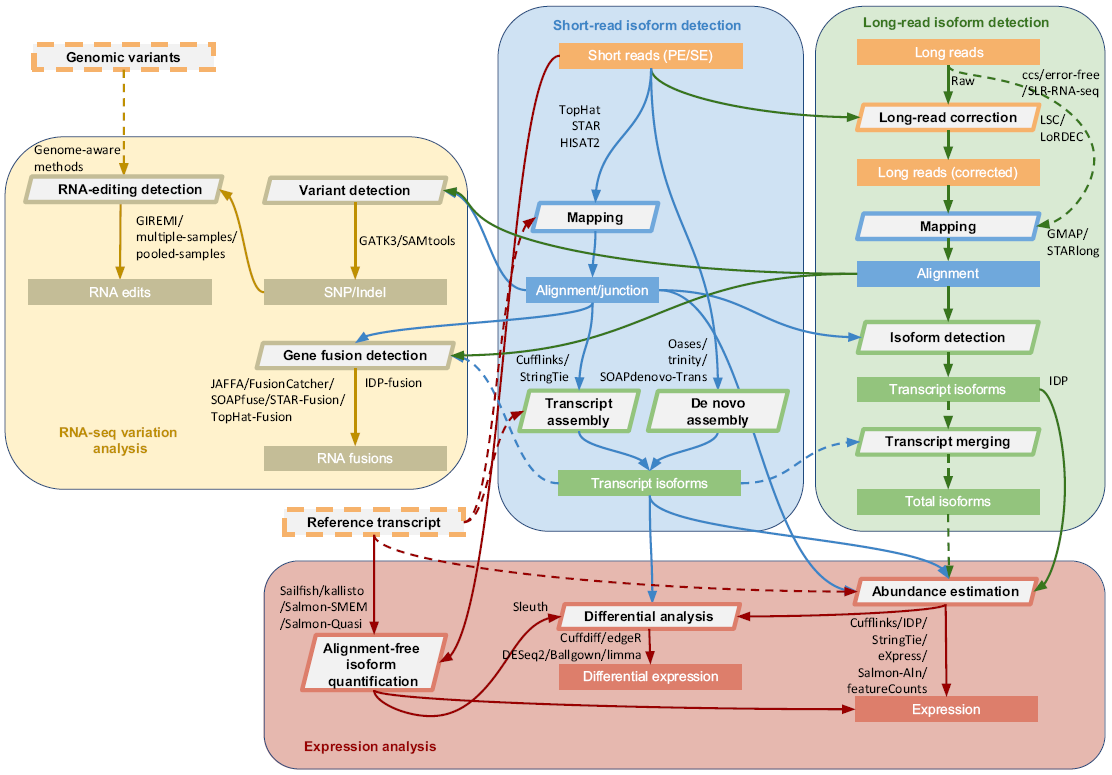
RNA-seq中最常用的分析方法就是找出差异表达基因(Differential gene expression, DEG)。在实验室中,标准流程就分为三步:
step1:构建测序文库。包括提取RNA,富集mRNA或清除核糖体RNA,合成cDNA,加上接头。
step2:在高通量测序平台(通常为Illumina)上对文库进行测序,每个样本的测序深度为10-30M读长。
step3:数据分析。具体而言:对测序得到的读长进行比对或组装到转录本上;对覆盖到每个基因区域的读长进行计数;根据统计模型鉴定不同样本间差异表达的基因。
(二) 差异基因表达的RNA-seq数据分析流程
主流的分析流程有三种,如下图分别用A、B、C三个方框表示的流程,虚线表示替代工具。
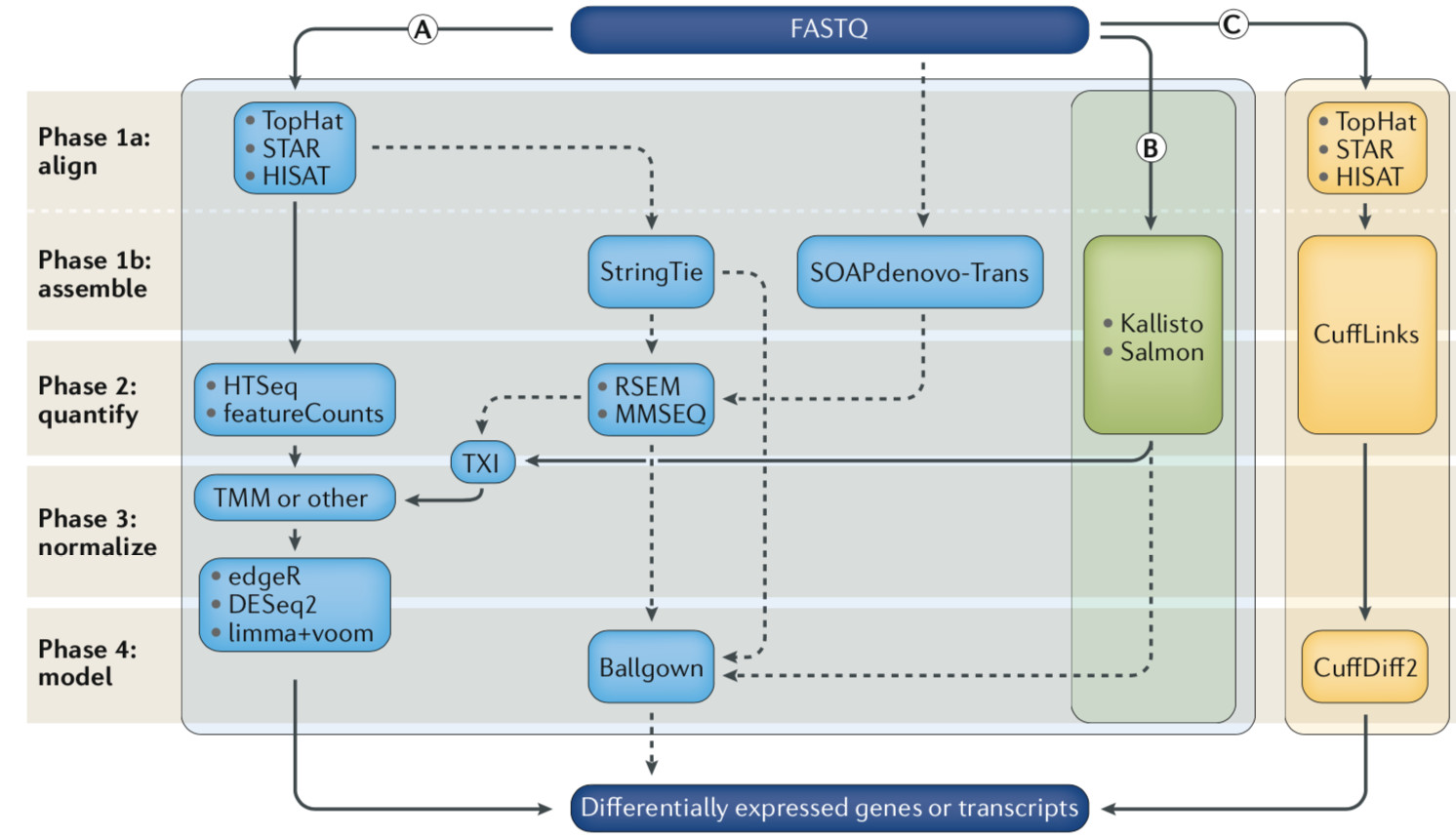
<1> 传统分析流程
上图的A分析流程是比较传统的分析方法,具体过程为:
- 通过比对工具如
TopHat、STAR或HISAT2等工具将测序读长数据比对回帖到参考基因组; - 然后使用一些定量工具如
HTSeq或featureCounts对比对到每个基因区域的读长进行计数; - 通过Excel或编程语言脚本产生一个列为样本、行为基因、填充数值为读长数目的基因表达矩阵;
- 为克服样本间测序深度的差异,需做TMM或FPKM等归一化处理;
- 通过
edgeR、DESeq2或limma+voom等统计工具进行差异表达分析,得到差异表达基因; - 进一步可视化或功能分析…
<2> 免比对分析流程
上图的B分析流程是较新的免比对分析方法,Kallisto 或 Salmon这些工具会在一步操作中组装转录本并对相应的转录本进行定量,然后通过与A分析流程中相同的归一化和统计分析,得到差异表达基因。
<3> 通过转录本估计丰度的分析流程
这种分流思路是先将测序数据比对到参考基因组,然后将短读长组装为转录本,最后估计转录本的丰度。例如在C分析流程中,首先于传统分析流程一样,需使用TopHat、STAR或HISAT2等工具将测序读长数据比对回帖到参考基因组;然后使用CuffLinks将短读长拼接成转录本;之后使用CuffDiff2来输出转录本丰度的估计值,以及一个差异表达基因或转录本的列表。
其它常用工具还包括StringTie,这个工具使用TopHat(或类似工具)的比对结果来组装一个转录本模型,然后利用RSEM 或 MMSEQ估计转录本丰度值。最后将转录本的丰度值输出给Ballgown来计算差异表达基因或转录本。
SOAPdenovo-trans这个工具则能同时对读长进行比对和组装,其结果输入给RSEM或MMSEQ以估计转录本丰度。
(三) 转录组分析工具比较
RNA-seq数据分析常用工具如下: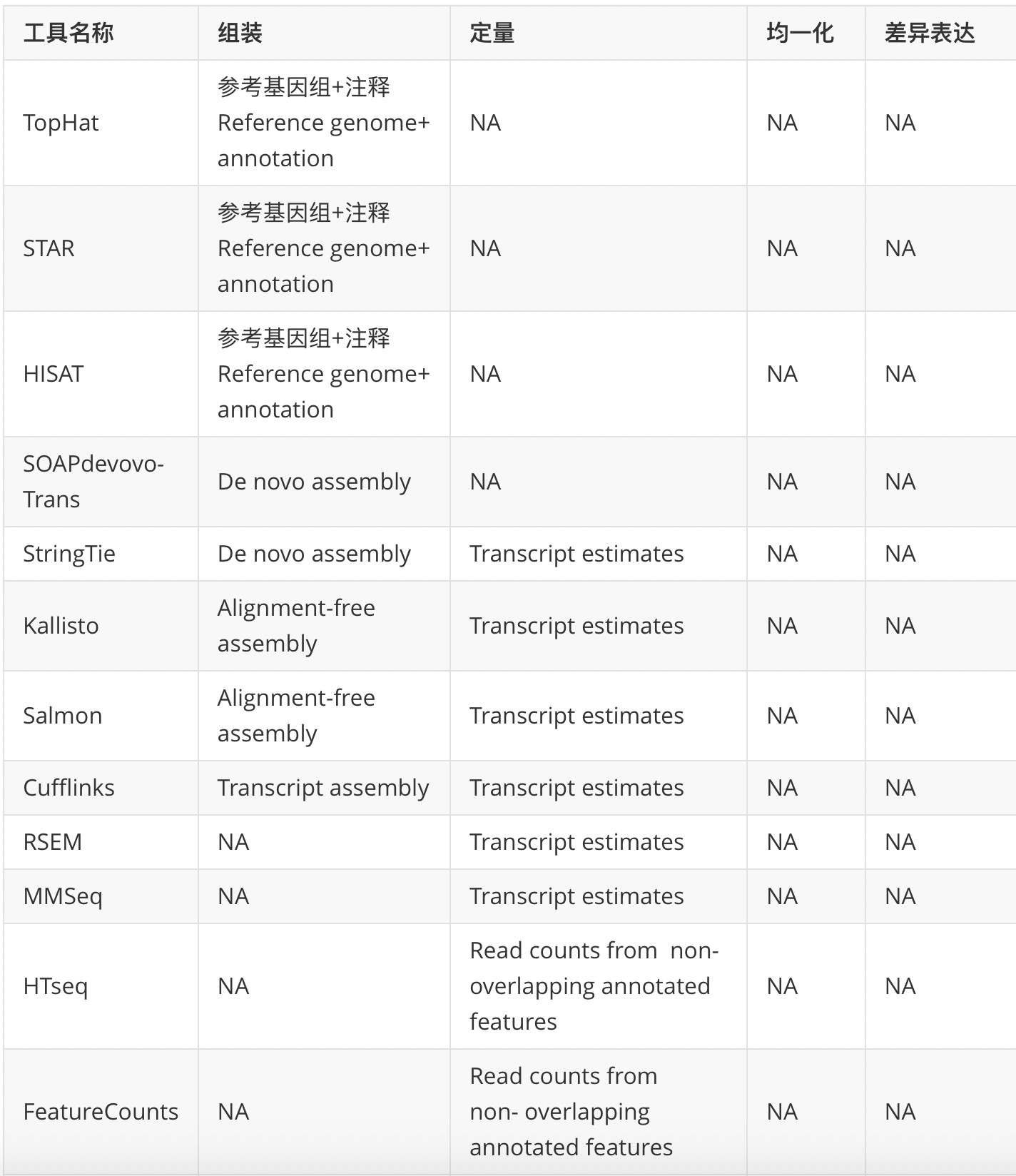
Nature Communication有一篇文章”Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis”对39个分析工具的120种常见组合方式进行了分析,总结了一套比较普适性流程。
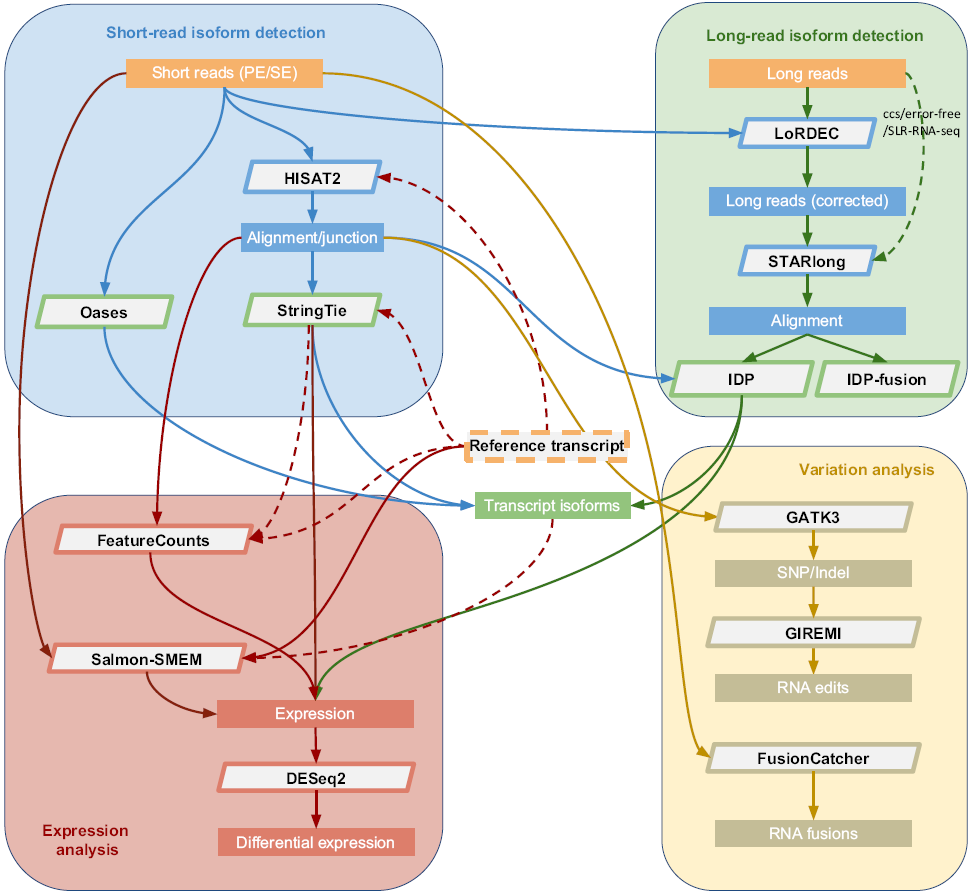
表达定量分析工具比较
不同分析工具的准确度和分析时间是有差异的,具体而言:
- HISAT2表现出最快的速度和最准确的拼接比对,但是敏感度不如STAR;
- StringTie在速度和准确性都优于Cufflinks;
- 免比对工具Salmon-SMEM和kallisto表现了最好的一致性和最高准确度,因此如果不是发现新的转录本,这两个工具可以作为快速而准确的方案。
- DESeq2和edgeR与免比对工具kallisto或Salmon-SMEM联用,可以获得高准确度的差异表达分析结果;
差异表达基因鉴定工具比较
有多种方法鉴定差异表达基因,包括基于计数 (reads count)的DESeq、limma和edgeR、基于组装技术的Cuffdiff和Ballgown、不经过比对定量进行差异分析的sleuth。
- DESeq2在所有组合中表现最佳,sleuth、edgeR和limma略微次之,但差别不大。
- Cuffdiff和Ballgown的准确度没有基于计数的工具准确度高。
- 基于计数的工具比基于组装的工具更高效, 免比对直接定量的工具如Salmon和kallisto能够获得高质量的差异分析结果。
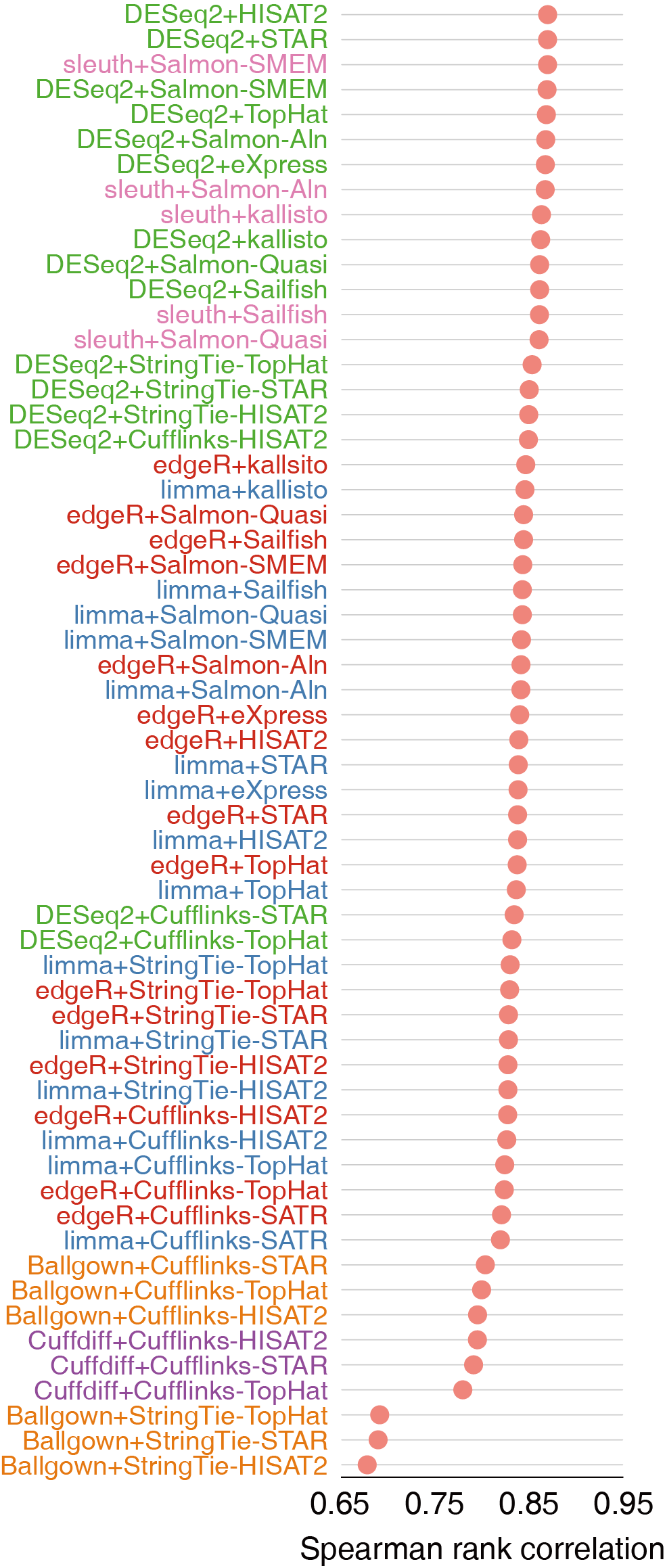
推荐使用的软件组合形式:
- 基于“比对 + 组装 + 定量”:
HISAT2:用于比对;StringTie用于转录本组装;RSEM用于估计转录本丰度;
- 基于“比对 + 计数 + 定量”:
HISAT2或STAR或TopHat:用于比对;DESeq2:用于定量
- 基于免比对:
Salmon-SMEM或kallisto:用于计数;sleuth或DESeq2:用于定量
###(四) 参考资料
原文作者: Billy & Barney
原文链接: https://liangbilin.github.io/2019/09/01/Billy--转录组差异表达分析概述/
版权声明: 转载请注明出处(必须保留作者署名及链接)