利用AutoDOCK进行分子对接
分子对接(Moleculer-docking)理论
分子对接就是两个或多个分子之间通过几何匹配和能量匹配相互识别找到最佳匹配模式的过程。分子对接在酶学研究和药物设计中具有重要的应用意义。
分子对接计算是在受体活性位点区域通过空间结构互补和能量最小化原则来搜寻配体与受体是否能产生相互作用以及它们之间的最佳结合模式。其思想起源于Fisher的锁钥模型(即一把钥匙开一把锁),主要强调的是空间形状的匹配。但配体和受体的识别要比这个模型更加复杂。首先,配体和受体在对接过程中会由于相互适应而产生构象的变化。其次,分子对接还要求能量匹配,对接过程中结合自由能的变化决定了两个分子是否能够结合以及结合的强度。
1958年D.E.Koshland提出分子识别过程中的诱导契合概念,受体分子活性中心的结构原本并非与底物完全吻合,但其是柔软和可塑的。当配体与受体相遇时,可诱导受体构象发生相应的变化,从而便于他们的结合进而引起相应的反应。
分子对接方法根据不同的简化程度分为三类:刚性对接、半柔性对接和柔性对接。刚性对接指在对接过程中,受体和配体的构象不发生变化,适合研究比较大的体系如蛋白-蛋白之间以及蛋白-核酸之间,计算简单,主要考虑对象之间的契合程度。半柔性对接常用于小分子和大分子的对接,在对接过程中,小分子的构象可以在一定范围内变化,但大分子是刚性的。这样既可以在一定程度上考察柔性的影响,又能保持较高的计算效率。在药物设计和虚拟筛选过程中一般采用半柔性的分子对接方法。柔性对接方法一般用于精确研究分子之间的识别情况,由于允许对接体系的构象变化,可以提高对接准确性但耗时较长。
分子对接的目的是找到底物分子和受体分子最佳结合位置及其结合强度,最终可以获得配体和受体的结合构象,但这样的构象可以有很多,一般认为自由能最小的构象存在的概率最高。搜寻最佳构象就要用到构象搜索方法,常用的有系统搜索法和非系统搜索法。系统搜索法通过改变每个扭转角评估所有可能的结合构象,进而选取能量最低的。这一方法计算量非常大。因此通常使用非系统搜索法来寻找能量较低构象,常用方法有:分子动力学方法、随机搜索、遗传算法、距离几何算法等。随机搜索又包括完全随机算法、蒙特卡罗法和模拟退火法等。
分子对接软件包——AutoDock
AutoDock软件简介
AutoDOCK是一款由Scripps研究所的Olson实验室开发与维护的开源的分子模拟软件,最主要应用于蛋白与小分子配体的分子对接。其用户图形化界面(GUI)工具为AutoDOCK Tools(ADT)。AutoDock的新一代产品为AutoDock Vina。
AutoDock Vina使用拉马克遗传算法提高效率。软件把遗传算法和局部搜索结合在一起,遗传算法用于全局搜索,而局部搜索用于能量优化。为了加快计算速度,AutoDock Vina采用格点(grid)计算。首先在受体活性氨基酸附近划定一个长方体区域作为搜索空间,扫描不同类型的原子计算格点能量,在搜索空间内,调整配体的构象、位置和方向,进而评分、排序获得能量最低的构象作为输出结果。
对范德华相互作用的计算:每个格点上保存的范德华能量的值的数目与要对接的配体上的原子类型数目相同。如果一个配体中含有C、H、O三种原子类型,那么每个葛店需要用单个探针原子与来计算其与受体之间的范德华相互作用值。当配体与受体进行分子对接时,配体中某个原子和受体之间的相互作用能通过周围8个格点上的这种原子类型为探针的格点值用内插法得到。
静电相互作用的计算采用静电势格点。当配体与受体对接时,某个原子和受体之间的静电相互作用能通过周围格点上静电势以及原子上的部分电荷计算得到。
准备docking需要的受体(蛋白)和配体(化合物)
Docking算法需要每个原子带有电荷并且需要标记原子的属性。这些信息通常未包含在PDB文件中。我们需要在对蛋白和小分子的PDB文件预处理,生成同时包含以上信息和PDB文件中原子坐标信息的PDBQT格式文件。进一步地对于“柔性配体docking”,我们还需要定义配体的柔性部分和刚性部分。所有这些都可以通过软件AutoDock Tools (adt)来完成。
准备受体蛋白
- PDB文件(1hsg.pdb)中包含了蛋白、配体和水分子;首先提取出蛋白的坐标,即以关键字ATOM和TER开头的行存储到文件1hsg_prot.pdb。
在Linux下,使用命令 egrep “^(ATOM|TER)” 1hsg.pdb > 1hsg_prot.pdb
- 启动AutoDockTools
选择支持GUI的客户端,如MobaXterm,连接服务器。进入AutoDockTools安装目录的’bin’子目录,使用命令 ./adt &
- 依次点选 File–Read Molecule–1hsg_prot.pdb加载蛋白分子。
ADT中按住左键拖动旋转分子结构;点击中键滚动缩放;按住右键移动晶体位置。
- 更改展示方式
依次点选 Color–By Atom Type–All Geometries–OK。
- 加氢:晶体结构中通常缺少氢原子的坐标(因为氢原子电子少,且质子核对电子吸引能力弱,因此很难定位,具体见 http://www.uh.edu/~chembi/ChemSocRev_Jones_critical.pdf )。但是在docking过程中,氢原子尤其是极性氢原子对计算静电作用是必须的。因此我们需要给蛋白加上氢原子,之后氢原子会以白色短线形式出现。
依次点选 Edit–Hydrogen–Add–Polar only–OK(只加极性H)

- 存储对蛋白的每个原子所做的修改和原子类型判断。
依次点选 Grid–Macromolecule–Choose–1HSG_protein–Slect Molecule (ADT会弹出一个信息框包含程序所做的处理,比如合并非极性氢原子,计算原子局部电荷和判断原子类型,并提示保存Save–1hsg_prot.pdbqt)
- 在受体蛋白定义配体结合的3D搜索空间。如果我们事先不知道结合位点,理论上可以定义一个长方体盒子包含整个蛋白或者随便一个特定区域 (下文PDB文件解析中会提到PDB文件中有时会包含活性位点信息)。
依次点选 Grid – Grid box ,将会在蛋白上画出一个长方体,并且有一个弹出框。在弹出框中,拖拽刻度线查看长方体的变化,完成设置。
在这个例子中,我们知道结合位点,就选取以其为中心的一个小空间。
设置Spacing (angstrom)为1埃 (这实际是一个换算系数, 相当于步长; 默认为0.375,是C-C单键长度的1/4,最大为1。spacing值与(各个维度上的点的数目+1)的乘积就是长方体Grid box的大小)。在我们调整的过程中,可以看到随着这个数值的变大,立方体也被放大了。
另外我们设置x,y,z center为16,25,4,number of points in (x,y,z)-dimension为30,30,30(最大为126,必须为偶数,AutoDock会自动再每一维再加一个点)。记下我们设置的这些点,下面会用到。在刻度转盘处点击右键会弹出一个窗口,输入数字回车即可设置GRID的中心坐标和大小。较大的number of points in (xyz)-dimension和较小的Spacing会增加搜索的精度,同时需要花费更多的计算时间。 - 设置受体的柔性残基:
在ADT中依次点选 Flexible Residues–Input–Choose Macromolecule–1hsg_prot; select–select from string–Residue: ARG8–Add–Dismiss, 8号ARG氨基酸残基就被选中了。
再依次点选Flexible Residues–Choose Torsions in Currently Selected Residues将选择的残基标记为柔性残基并设置可扭转的数量。
在分子显示窗口中分别点击两个残基上CA和CB原子之间的建,使之变为非扭转的(紫色显示)[Shift + 鼠标左键],这样两个残基中的32个键中有6个是可扭转的。这里设置配体的柔性残基或者使CA-CB的键为刚性都是可选操作。
放在教程中只是用来展示怎么操作的,无其它指导意义。Flexible Residues–Output–Save Flexible PDBQT保存柔性残基文件。 Flexible Residues–Output–Save Rigid PDBQT保存柔性残基文件。 - 关掉grid和删除protein
Grid Options–File–Close w/out saving; Edit–Delete–Delete Molecule–1hsg_prot–Continue
准备配体
与蛋白结构类似,配体的结构也缺少氢原子,我们需要添加氢原子并且定义哪些键是可以旋转的以用于柔性docking。
- 从PDB结构中提取配体的原子位置。indinavir的配体残基名字为MK1,以HETATM开头的行表示非核心多聚体的成分 (heteroatoms)(具体见PDB文件格式解释)。
Linux系统下,运行 grep “^HETATM.*MK1” 1hsg.pdb > indinavir.pdb
- 将结构读入ADT:
依次点选 File–Read Molecule–indinavir.pdb
依次点选 Color–By Atom Type–All Geometreies–OK - 给配体加上氢原子:
Edit–Hydrogen–Add–Polar only–OK
- 在ADT中定义此化合物为配体,以便ADT为其计算局部电荷(partial charges)和设置可旋转配体键。
依次点选 Ligand–input–Choose–indinavir–Select Molecule for AutoDock4
这时会有一个弹出框显示ADT所做的操作,包括合并非极性氢(只在添加了的情况下)、计算电荷电量和设置旋转键。
然后点选 Ligand–Output–Save as PDBQT 存储结果。 - 查看ADT检测出的旋转键:
依次点选 Ligand–Torsion Tree–Choose Torsions,可以看到Number of rotatable bonds=14/32。
准备docking配置文件
docking配置文件包含了输入的受体(蛋白)、配体(化合物)和搜索参数的信息,为一个文本文件,名字任意,可以为conf.txt,内容如下:
receptor = 1hsg_prot.pdbqt
ligand = indinavir.pdbqt
num_modes = 50
out = dockingResult.pdbqt
log = docking.log
center_x = 16
center_y = 25
center_z = 4
size_x = 30
size_y = 30
size_z = 30
seed = 2009
receptor和ligand为输入文件的名字,与conf.txt在同一目录下; out为输出文件的名字;log为输出日志文件的名字。
centerhe和size定义搜索空间的位置和大小。
num_modes设置最多显示的结合模型,鉴于只输出符合能量值要求的结果,最后输出的结合模型数量可能少于这一数值。
seed设置随机数生成的种子,可以为任意整数。如果想重现之前的分析结果就需要使用相同的seed。
Docking 小分子化合物indinavir到HIV-1蛋白酶
- 使用AutoDock Vina执行docking预测
在Linux终端执行 path_to_vina/vina –config conf.txt
- 输出结果包含两个文件,构象文件dockingResult.pdbqt和日志文件docking.log.
dockingResult.pdbqt: 包含所有docking的模式,通常第一个为结合最好的构象,但如果前几个能量值相差不大时也有例外。
docking.log: 日志文件,包含结合能量值(第一列,越低越稳定,默认由低到高排序,所以第一个为最好的构象)、每个构象与第一个构象的距离、每个构象与第一个构象的差别。
用Pymol可视化结果
打开PyMOL,依次点选File–Open文件类型选择All Files-选取结果dockingResult.pdbqt文件、原始蛋白和配体的pdb文件、原教程的pdbqt文件。
用Pymol展示配体和受体相互作用的原子和氢键
为了简化展示过程,我们设计了一个pml脚本 (脚本内有很详细的解释),只需要修改脚本里面受体和配体的名字,然后在PyMOL的命令行界面输入PyMOL> run display.pml即可获得展示结果。当然这个脚本也可以使用程序generatePmlForHbond.py生成。
1 | ############################################################ |
输出结果如下:
PyMOL>run E:/docking/list_hbonds.py
B/MK1’902/N4 B/GLY’27/O 3.03
B/MK1’902/O4 B/GLY’27/O 3.16
B/MK1’902/O2 A/ASP’25/OD1 2.77
B/MK1’902/O2 B/ASP’25/OD1 2.63
看上去比显示的氢键少了三个,这是因为我们在第二个函数中使用了H-键角度限制,如果在调用时给定参数list_hb(mode=0)则会获得一致结果。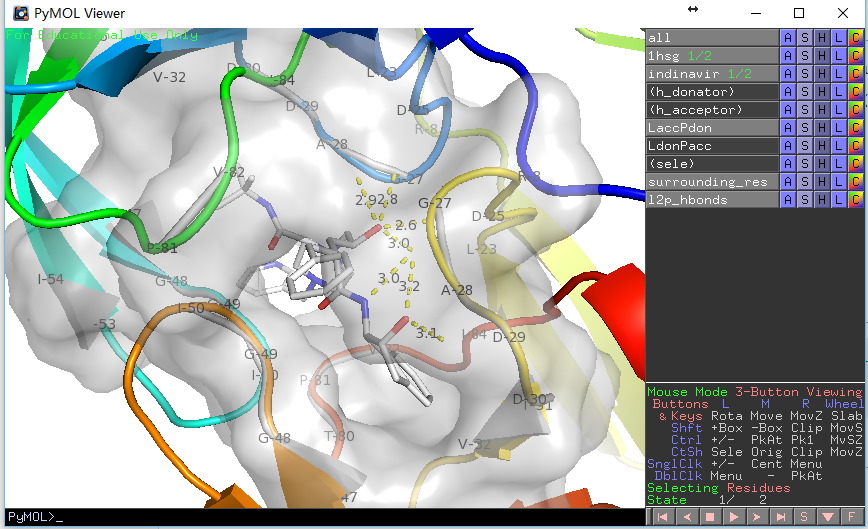
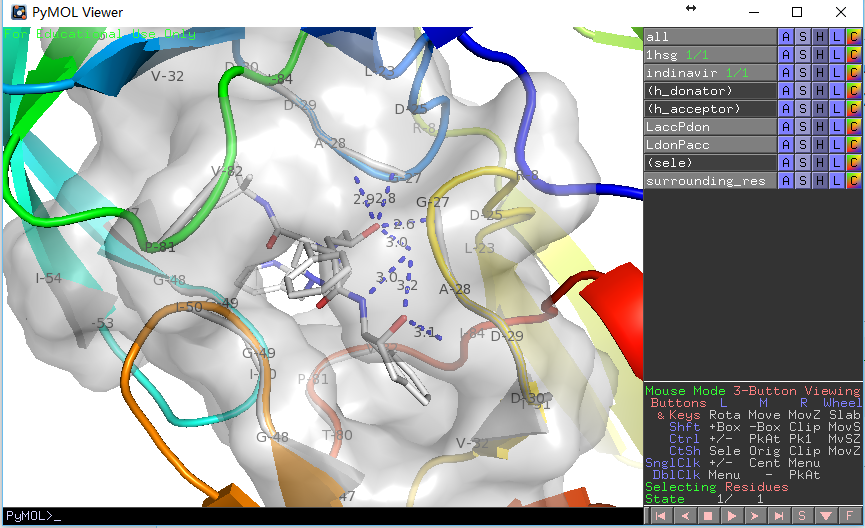
H-bond结果展示。第一张图为运行display.pml后的结果,蓝色虚线为氢键;第二张图为运行list_hbonds.py后的结果, 黄色虚线为氢键(覆盖了之前的蓝色)。可以通过点选LaccPdon, LdonPacc, l2p_hbonds显示不同的氢键。
展示疏水表面
1 | # color_h |
将上面的脚本存储为color_h.py,在PyMOL界面运行File–Run–color_h.py,在命令行输入
PyMOl color_h –Show surface
原文作者: Billy & Barney
原文链接: https://liangbilin.github.io/2019/07/25/Billy--利用AutoDOCK进行分子对接/
版权声明: 转载请注明出处(必须保留作者署名及链接)